Genetic determinants for virulence and adaptation of clade 2.3.4.4 H5N8 viruses in chickens, ducks and mammals

David Scheibner, Claudia Blaurock, Angele Breithaupt, Thomas C. Mettenleiter and Elsayed M. Abdelwhab
Abstract
From 2014 to 2021 the panzootic H5N8 clade 2.3.4.4 AIV spread on an unprecedented scale to wild and domestic birds worldwide. Compared to the parental clade 2.3.4.4A viruses (designated H5N8-A) in 2013/2015, viruses in clade 2.3.4.4B (designated H5N8-B) from 2015 to 2021 caused high mortality in wild birds and Pekin ducks and were detected recently in mammals. In this comprehensive study, we analyzed pathogenicity and virulence markers of H5N8-A and H5N8-B in chickens, ducks and mice. Recombinant H5N8-A, H5N8-B and H5N8-A viruses carrying gene segment(s) from H5N8-B were successfully constructed using reverse genetics. In vitro, compared to H5N8-A, H5N8-B virus exhibited increased binding affinity to avian-sialic acid receptors and higher NA activity. Both H5N8-A and H5N8-B blocked IFN-β induction efficiently despite the striking preferential differences in the NS1 C-terminus domain (CTE). In chickens, both viruses were highly virulent, while H5N8-B was more virulent in Pekin ducks and mice than H5N8-A. In ducks, HA, NA and NS played a role in H5N8-B virulence and transmission. Moreover, we found that mutations in the NS1 CTE of H5N8-B was crucia for virus adaptation in mammals.
Talk at the Junior Scientist Symposium 2022, Greifswald, Germany (14 - 16 November, 2022)
Virological and epidemiological aspects of the spillover of HPAIV Gs/Gd H5 from poultry into wild birds

Thijs Kuiken
Talk at the JNCC-BTO UK Workshop on HPAI and wild birds, online (10 November, 2022)
Losing the upper hand: Establishment of highly pathogenic avian influenza in wildlife

Thijs Kuiken
Abstract
Highly pathogenic avian influenza (HPAI) typically emerges in commercial poultry farms by conversion of an innocuous wild-type virus—aptly called low pathogenic avian influenza virus—into one that causes high mortality in poultry. The current H5N1 HPAI virus—clade 2.3.4.4b of the A/Goose/Guangdong/1/96 lineage—originated in a commercial goose farm in China in 1996 and spread across the rapidly growing poultry populations in Asia, substantially spilling over into wild birds in 2005. The virus caused numerous wild bird outbreaks, typically during autumn and winter, but has persisted year-round in wild birds in Europe since 2021. This year, it has spread quickly to breeding seabirds in Europe, North America, and Africa. For many of these long-lived species, already threatened by loss of habitat and climate change, the resulting mortality will have a large impact on their populations. Short-term recommendations include accurate monitoring of the virus and associated mortality in wild birds, and, where appropriate, coordinated removal of infected wild bird carcasses to limit virus spread. Long-term recommendations include enhanced protection of seabird and waterbird sites, vaccination of poultry against HPAI, reduction of poultry farm size and density, and avoidance of waterbird-rich areas as a location for poultry farms. The ongoing global spread of HPAI H5 viruses stresses the importance of international cooperation to better understand the global epidemiology of avian influenza, and is a call to re-assess the poultry sector in a way that embraces the One Health perspective: to sustainably balance and optimize the health of people, animals and ecosystems.
Talk at the BWDS DSWH joint conference, Utrecht, The Netherlands (13 October, 2022)
Using scenario tree modelling for targeted flock sampling to substantiate freedom from disease after the 2021 22 HPAI epidemic in Italy

Scolamacchia F., Mulatti P., Fornasiero D., Dorotea T., Manca G., Mannelli A.
Poster at the ECVPH AGM & Annual Scientific Conference 2022, Athens, Greece (28-30 September, 2022)
Neuraminidase stalk deletion decreased virulence and/or transmission of avian influenza viruses H5N8 and H7N7 in poultry

David Scheibner, Anne Dittrich, Claudia Blaurock, Thomas C. Mettenleiter, Elsayed M. Abdelwhab
Abstract
Avian influenza viruses (AIV), members of the RNA family Orthomyxoviridae, infect a wide range of wild and terrestrial birds. In wild waterfowl, the natural reservoir, AIV are thought to be in a static evolution; however, the transmission of AIV to poultry is accompanied by mutations in different viral proteins including the neuraminidase (NA). NA is a surface glycoprotein and varies into 9 subtypes (N1-N9). It is formed of stalk and head domains and mediates virus release from infected cells. Deletions in the NA stalk domain (NAdel) particularly in H5N1 viruses have been observed and linked to increased virus adaptation and/or virulence in chickens. Here, we investigated the impact of NA deletion or extension on biological fitness of two other NA subtypes: high pathogenic H5N8 (2016), with naturally full NA stalk and low pathogenic H7N7 (2011) with a natural NA stalk deletion. Using reverse genetics, we generated an H5N8 with a deletion in the stalk domain and another H7N7 with NA stalk extension. The replication of these four viruses in experimentally inoculated chickens and ducks was studied. In vitro data revealed no significant impact on virus replication on avian or mammalian cells. In contrast, reduction in the NA activity after elongation of the H7N7 NA stalk could be shown. In vivo, viruses carrying full NA stalk domain exhibited higher virulence and more efficient replication in chickens and ducks. These results indicate that the NA stalk deletion affects virus fitness in vivo and in vitro.
Talk at the 2nd Summer School "Infection Biology" 2022, Greifswald, Germany (26 - 28 September, 2022)
Different genetic constellations for high virulence, transmission and tropism of highly pathogenic H7N7 avian influenza virus in turkeys and chickens

Scheibner, David; Ulrich, Reiner G.; Mettenleiter, Thomas C.; Abdelwhab, El-Sayed M.
Abstract
Highly pathogenic (HP) avian influenza viruses (AIV) cause severe mortality in chickens (Gallus gallus domesticus) and turkeys (Meleagris gallopavo), although turkeys are more sensitive than chickens. Little is known about the virulence determinants in both bird species, beyond the polybasic cleavage site (CS) in the hemagglutinin (HA). In 2015, HPAIV H7N7 and a putative low pathogenic (LP) ancestor were isolated from the same chicken farm. In addition to the polybasic HA CS, mutations in all gene segments were observed. The aim of this study was to investigate the impact of mutations in different segments in addition to the polybasic CS on virulence, transmission, replication and tissue tropism in chickens and turkeys. Using reverse genetics different virus reassortants with an LP backbone carrying the polybasic CS and single or multiple HP segments were generated. Interestingly, viruses carrying the HP NS or M gene segments or NS segment were as virulent and transmissible as the HPAIV in turkeys and chickens, respectively. All viruses were detected in the brain, although the endotheliotropism was exclusively found in chickens, where the HACS and to lesser extent NA are major determinants for the endotheliotropism. In vitro, HP exhibited lower NA and higher polymerase activities compared to LP. Taken together, this study showed two major differences between chickens and turkeys: more genetic constellations to confer high virulence and transmission in turkeys than in chickens and neurotropism in turkeys vs. endothelial tropism in chickens.
Poster at the 2nd Summer School "Infection Biology" 2022, Greifswald, Germany (26 - 28 September, 2022)
Genetic determinants for virulence and adaptation of clade 2.3.4.4 H5N8 viruses in chickens, ducks and mammals
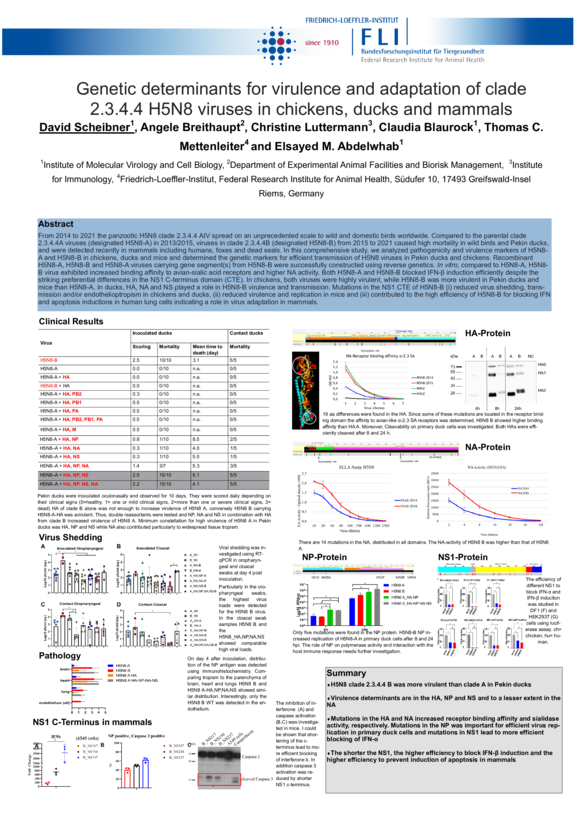
David Scheibner, Claudia Blaurock, Angele Breithaupt, Christine Luttermann, Thomas C. Mettenleiter and Elsayed M. Abdelwhab
Abstract
From 2014 to 2021 the panzootic H5N8 clade 2.3.4.4 AIV spread on an unprecedented scale to wild and domestic birds worldwide. Compared to the parental clade 2.3.4.4A viruses (designated H5N8-A) in 2013/2015, viruses in clade 2.3.4.4B (designated H5N8-B) from 2015 to 2021 caused high mortality in wild birds and Pekin ducks, and were detected recently in mammals including humans, foxes and dead seals. In this comprehensive study, we analyzed pathogenicity and virulence markers of H5N8-A and H5N8-B in chickens, ducks and mice and determined the genetic markers for efficient transmission of H5N8 viruses in Pekin ducks and chickens. Recombinant H5N8-A, H5N8-B and H5N8-A viruses carrying gene segment(s) from H5N8-B were successfully constructed using reverse genetics. In vitro, compared to H5N8-A, H5N8-B virus exhibited increased binding affinity to avian-sialic acid receptors and higher NA activity. Both H5N8-A and H5N8-B blocked IFN-β induction efficiently despite the striking preferential differences in the NS1 C-terminus domain (CTE). In chickens, both viruses were highly virulent, while H5N8-B was more virulent in Pekin ducks and mice than H5N8-A. In ducks, HA, NA and NS played a role in H5N8-B virulence and transmission. Mutations in the NS1 CTE of H5N8-B (i) reduced virus shedding, transmission and/or endothelioptropism in chickens and ducks, (ii) reduced virulence and replication in mice and (iii) contributed to the high efficiency of H5N8-B for blocking IFN and apoptosis inductions in human lung cells indicating a role in virus adaptation in mammals.
Poster at the 2nd Summer School "Infection Biology" 2022, Greifswald, Germany (26 - 28 September, 2022)
The interplay between avian influenza viruses and their hosts: insights from transcriptomic sequencing of galliformes infected with low pathogenic viruses of the H7 subtype

Gianpiero Zamperin, Alice Bianco, Jacqueline Smith, Alessio Bortolami, Lonneke Vervelde, Alessia Schivo, Andrea Fortin, Sabrina Marciano, Valentina Maria Panzarin, Eva Mazzetto, Adelaide Milani, Yohannes Berhane, Paul Digard, Francesco Bonfante, Isabella Monne
Abstract
Background
Influenza A viruses (AIVs) are important human and veterinary health pathogens, with avian species representing their natural hosts. There are many subtypes of AIVs, each characterized by a specific pair of surface glycoproteins, the hemagglutinin (HA) and the neuraminidase (NA). AIVs infecting poultry can be divided into two groups, according to the severity of the disease they cause. So far, only H5 and H7 subtypes have showed the capability to mutate from low to highly pathogenic avian influenza (LPAI, HPAI). Once this happens, the mutation results in severe epizootics with up to 100% mortality. Although AIVs are inextricably linked to their hosts in their evolutionary history, the contribution of host-related factors in the emergence of HPAI viruses has only been marginally explored.
Methods
We selected two pairs of H7 low-pathogenic viruses with a distinctive ability to evolve from LPAI to HPAI under natural conditions. One pair of LPAI virus precursor of HPAI strains (precHP) included the H7N1 A/chicken/Italy/1279/1999 and the H7N3 A/chicken/BC/CN006/2004. The other pair included two viruses that have not evolved into highly pathogenic strains (nevoLP), even after mid- to long-term circulation in Galliformes: the H7N3 A/turkey/Italy/2962/03 and H7N2 A/chicken/Italy/1670/15. We did an in vivo infection of six-week-old female SPF white leghorn chickens. RNA samples were isolated from tracheas at 24, 36, 48 at 72 hours post-infection and subjected to library production and sequencing. High quality data were aligned to host genome to perform differential expression analysis. Differentially expressed genes (FDR < 0.05, |log2FC| ≥ 1) were used for GO enrichment analysis (FDR < 0.05) and pathway analysis with IPA.
Results
NevoLP enriched terms included chromosome segregation, cell division, cell cycle, biosynthetic process, cellular component biogenesis, ATP metabolic process, cellular metabolic process, suggesting that infected cells were busy multiplying and producing proteins, ATP and other components. PrecHP enriched terms included immune response, immune effector process, response to external stimulus, response to biotic stimulus and actin filament-based process, indicating a host immune response as the main reaction to the viral infection.
Conclusion
Our challenge study demonstrated that the number and type of DEGs can profoundly vary between viruses and even within the same HA subtype, pathotype, species and tissue. It is interesting to highlight that both the H7 HPAI precursors showed a very high number of general enriched terms related to immune response. These findings suggest that an exacerbated innate immune response may represent a selective factor shaping AIV pathogenicity evolution.
Poster at the OPTIONS XI for the Control of INFLUENZA Conference, Belfast, UK (26-29 September, 2022)
Highly pathogenic avian influenza H5N1/H9N2 reassortant virus in West Africa: a potential threat for humans?

Lalidia Bruno Ouoba, Habibata Lamouni Zerbo Ouermi, Bianca Zecchin, Giacomo Barbierato, Sandaogo Hamidou Ouandaogo, Elisa Palumbo, Edoardo Giussani, Alessio Bortolami, Calogero
Terregino, Mariétou Guitti Kindo, Dominique Guigma, Nicolas Barro, Alice Fusaro , Isabella Monne
Abstract
Background
Since 2006, the West African poultry population has been seriously hit by different waves of highly pathogenic avian influenza (HPAI) H5Nx viruses, brought by migratory birds, with the last introduction of the H5N1 subtype reported in late 2020. In January 2017, the H9N2 subtype of the G1 lineage was identified for the first time in Burkina Faso and since then it has been reported in several West African countries. The co-circulation of these two zoonotic subtypes in this region is a cause for concern not only for animal health and economic damage, but also for the possible emergence of reassortant viruses with unknown biological properties and public health implications.
In December 2021, three HPAI H5N1 outbreaks were reported in poultry farms from three distinct regions of Burkina Faso. Here we describe the complete genome characterization of the first H5N1/H9N2 reassortant virus in West Africa.
Methods
By using an Illumina MiSeq platform, we generated the whole genome sequences of three H5N1 viruses from Burkina Faso. Maximum likelihood phylogenetic trees were obtained for all the eight gene segments using IQTree v1.6.6 and the time to the most recent common ancestor (tMRCA) was estimated for the hemagglutinin (HA) gene using the BEAST v.1.10.4 package.
Results
Phylogenetic analysis of the HA gene shows that the HPAI H5N1 viruses from the three distinct outbreaks in Burkina Faso cluster together (similarity of 99%-99.2%) within clade 2.3.4.4B, and are closely related to HPAI H5N1 viruses identified in Nigeria and Niger in 2021-2022. The phylogenetic trees of the other gene segments confirm this clustering, except for the PA gene where the three H5N1 viruses from Burkina Faso cluster with H9N2 viruses collected in West Africa between 2017 and 2020. The tMRCA of the three H5N1 under study was June 2021 (95% HPD March-August 2021).
These reassortant viruses possess several mutations that may be associated to an increased zoonotic potential, including the HA-S137A (H3 numbering) mutation, which has been demonstrated to increase virus binding to human receptors, and the PA-367K mutation, which has been associated to a more efficient H5N1 virus replication in primary human bronchial epithelial (NHBE) cells, in A549 and Calu-3 cells.
Conclusion
Although it is difficult to ascertain where and when the reassortment event occurred, the emergence of an H5N1/H9N2 reassortant virus in West Africa raises concerns about its possible impact on animal and human health. Further studies to evaluate the biological significance of this emerging genotype and to assess its spread are urgently required.
Poster at the OPTIONS XI for the Control of INFLUENZA Conference, Belfast, UK (26-29 September, 2022)
TRANSIENT STEM-LOOP RNA STRUCTURES AND PURINE-RICH SEQUENCES AT THE H5 HEMAGGLUTININ CLEAVAGE SITE DRIVE DUPLICATIONS BY THE INFLUENZA POLYMERASE: A POTENTIAL EXPLANATION OF HIGHLY PATHOGENIC AVIAN INFLUENZA GENESIS

Mathis Funk, Monique I. Spronken, Anja C. de Bruin, Aartjan J.W. te Velthuis, Alexander P. Gultyaev, Ron A.M. Fouchier, Mathilde R. Richard
Poster at the OPTIONS XI for the Control of INFLUENZA Conference, Belfast, UK (26-29 September, 2022)
Genetic investigation of the HPAI H5N1 viruses responsible of HPAI epidemic in Italy in 2021 2022

Bianca Zecchin, Alice Fusaro , Giacomo Barbierato, Edoardo Giussani, Diletta Fornasiero, Francesca Scolamacchia, Paolo Mulatti, Annalisa Salviato, Alessia Sc hivo, Elisa Palumbo, Maria Varotto, Federica Gobbo, Isabella Monne, Calogero Terregino
Abstract
The last two epidemic waves caused by the H5Nx Highly Pathogenic Avian Influenza (HPAI) viruses of clade 2.3.4.4b occurring in Europe in 2020-2021 and 2021-2022 had devastating economic impacts on the poultry industry of several countries. Between October 2021 and April 2022, Italy reported 317 HPAI H5N1 outbreaks in poultry, which caused the death of 13'700'000 domestic birds. Most of the outbreaks occurred in the highly density poultry populated areas of northern Italy. To shed light on the number of introductions and diffusion dynamics of the H5N1 in Italy, evolutionary analyses (BEAST v1.10.4, Network10, IQtree v1.6.6) were performed on the complete genomes of 342 viruses identified in poultry (N=320) and wild birds (N=22), generated using an Illumina MiSeq platform.
Our analysis revealed at least twelve distinct introductions (seven in domestic birds) which had been caused by six distinct genotypes originating from different reassortment events. We observed a clear clustering by province, which suggested the occurrence of several secondary outbreaks. The Bayesian phylogeographic analysis suggested that the provinces of Verona and Mantua (north eastern Italy) had been the main source of the virus for the other provinces. Furthermore, the Bayesian analyses showing the species contribution to the viral spread revealed that, among the infected species, turkeys acted as the most likely source of the virus for the other domestic birds. Some mutations associated with an increased binding to human-type receptors were detected in the HA protein of all (S137A, H3 numbering) or few (S159N/T160A, H3 numbering) viruses. Besides, two mutations associated to an increased polymerase activity in mammalian cells (NP-N319K and PB2-K482R) were detected in a H5N1 virus from a wild bird.
Our genetic investigation of the HPAI H5N1 viruses responsible of the 2021-2022 Italian epidemic revealed the incursions of multiple viral genotypes. Nevertheless, a single genotype accounted for most of the outbreaks in poultry. Mutations associated with mammalian adaptation were only sporadically identified in the analysed viruses from avian hosts. However, the persistent circulation and continuous emergence of new reassortant viruses in Italy and Europe raise concern for animal and public health.
Poster at the 12th ESVV International Congress, Ghent, Belgium (20-23 September, 2022)
Going global: the ongoing adaptation of the Goose/Guangdong lineage of highly pathogenic avian influenza virus to wild birds

Thijs Kuiken
Abstract
The A/Goose/Guangdong/1/96 (GsGd) lineage of highly pathogenic avian influenza (HPAI) H5 virus first emerged in poultry in Southeast Asia more than 25 years ago. For the first time in 2005, the virus spread from Asia to Russia, western Europe, Africa and the Middle East, causing high mortality in wild birds and poultry. This spread was a result of unprecedented long-distance transport of HPAI virus, in which wild migratory ducks, geese and swans were implicated. Since then, HPAI GsGd H5 outbreaks have frequently occurred in Europe. In December 2021, H5N1 HPAI viruses were detected in poultry and a free-living gull in St. John’s, Newfoundland and Labrador, Canada. Our phylogenetic analysis showed that these viruses were most closely related to HPAI GsGd viruses circulating in northwestern Europe in spring 2021. Our analysis of wild bird migration suggested that these viruses may have been carried across the Atlantic via Iceland, Greenland/Arctic or pelagic routes. The unusually high presence of the viruses in European wild bird populations in late winter and spring 2021, as well as the greater involvement of barnacle and greylag geese in the epidemiology of HPAI in Europe since October 2020, may explain why spread to Newfoundland happened this winter (2021/2022), and not in the previous winters. To prevent and mitigate the risk of viral spread, it will be vital to further increase surveillance of wild birds in North America and South America, as well as at migration stop-over stations in Iceland and Greenland. The ongoing global spread of HPAI H5 viruses stresses the importance of close international cooperation and data exchange to better understand the global epidemiology of avian influenza, and is a call to re-assess the poultry sector in a way that embraces the One Health perspective: to sustainably balance and optimize the health of people, animals and ecosystems.
Talk at the 12th ESVV International Congress, Ghent, Belgium (20-23 September, 2022)
On ducks and flu – avian influenza in migratory waterfowl

Jonas Waldenström
Talk at the 6TH PAN-EUROPEAN DUCK SYMPOSIUM, Coimbra, Portugal (19-23 September, 2022)
Highly pathogenic avian influenza virus genesis and emergence

Mathilde Richard
Lecture at the EpiViral summer school, Alveiro, Portugal (09 September, 2022)
Striking variations in pathogenesis and virulence determinants of H7N1 in two closely related galliforms

Angele Breithaupt, Claudia Blaurock, Florian Pfaff, Thomas C. Mettenleiter and Elsayed M. Abdelwhab
Abstract
In gallinaceous birds, chickens and turkeys, the transition of low pathogenic (LP) avian influenza virus (AIV) of H5 and H7 subtypes to highly pathogenic (HP) AIV is accompanied mainly by changing the hemagglutinin (HA) monobasic cleavage site (CS) to a polybasic motif (pCS). For unknown reasons, turkeys are succumbed to higher morbidity and mortality to HPAIV-infection than chickens. Here, we determined the pathogenesis of H7N1 virus using recombinant Lp, Hp and Lp H7N1 carrying pCS (Lp_poly) in turkeys and chickens, and explored the host responses in this species compared to chickens. We found that recombinant Lp_poly was avirulent in chickens, but exhibited high virulence in turkeys indicating that virulence determinants vary in these two galliform species. Strikingly, the virulence of Hp in chickens and turkeys was associated with extensive tropism to the endothelium, pCS conferred endotheliotropism only in turkeys. Transcriptome analysis indicated that turkeys mount a different host response (e.g. RSAD2, USP18) than chickens (e.g. Mx1) particularly from genes involved in RNA metabolism and immune response. Together, these findings indicate variable pathogenesis, virulence determinants and host response in two closely related galliformes, turkeys and chickens, after the infection with HPAIV H7N1 and may explain the high vulnerability of turkeys to HPAIV.
Poster at the 8th International Influenza Meeting 2022, Muenster, Germany (2-4 September, 2022)
Recent changes in the epidemiology of the H5NX Goose/Guangdong lineage of HPAI virus

Thijs Kuiken
Talk at the 15th International Seabird Group Conference, Cork, Ireland (22 August, 2022)
Epidemiological investigations and analyses of H5N1 HPAI outbreaks in north Italy in 2021-2022

Diletta Fornasiero, Tiziano Dorotea, Bianca Zecchin, Alice Fusaro, Matteo Mazzucato, Paolo Mulatti
Abstract
Objective(s)
From October 2021, Italy has been involved in a severe highly pathogenic avian influenza (HPAI) epidemic sustained by a H5N1 virus. As of February 2021, 309 outbreaks in poultry were notified mainly in the north-eastern Regions. Despite enhanced biosecurity and surveillance measures, the epidemic spread unprecedentedly fast, with massive economic impacts. We present a preliminary assessment of the potential reasons leading to such rapid spread, accounting for both epidemiological evidences and phylogenetic support.
Materials and methods Epidemiological investigations mainly aimed at identifying risk factors and contacts between infected and non-infected farms occurring during a temporal risk window. Investigated contacts included: movements of vehicles/personnel, sharing of equipment, and/or belonging to the same owners.
These data where then integrated with genetic information of the complete genome of 298 viruses, and geographical distances between outbreaks, to assess possible scenarios of the epidemic evolution, hence granting insights in the disease dynamics.
Results
Preliminary phylogenetic analyses indicated at least 7 new introductions from wild to domestic birds, while contact-tracing activities showed 37 at-risk contacts related to vehicles/personnel movements, and 65 outbreaks belonging to the same owners. The close proximity of farms in such densely populated poultry areas, and at-risk contacts appear to be the main factors driving the rapid spread of the disease, leading to several secondary outbreaks. The phylogenetic and network analyses based on complete genome pinpointed that other potential unregistered and uncontrolled contacts between outbreaks occurred.
Conclusion
The integration of different analyses permits to monitor the epidemic dynamic, allowing real-time interventions to re-modulate the implementation of control measures. The recent epidemic demonstrates the need to further strengthen the biosecurity measures, considering the limited possibility of reducing the farms density, and plausibly the reassessment of the poultry sector organization to contain the risk of future epidemics.
Talk at the 16th International Symposium of Veterinary Epidemiology and Economics, Halifax, Canada (7-12 August, 2022)
Development of a Support Decision System dedicated to the management of Highly Pathogenic Avian Influenza epidemic in Italy.

Matteo Mazzucato, Paola Bonato, Monica Lorenzetto, Matteo Trolese, Grazia Manca, Nicola Ferrè
Abstract
Objective(s)
From October 2021, North-eastern Italy was involved in a severe H5N1 High Pathogenic Avian Influenza epidemic. In less than three months, more than 300 outbreaks were notified with massive repercussions on the poultry production. The high number of daily cases hampered the capacity to counter the disease spread, prompting for the development of a tool to keep track of the evolving epidemiological situation and control measures applied. Therefore, a Support Decision System was developed, exploiting well-established data interoperability standards, data warehouse framework, and web-based GIS technologies. This led to an integrated system for visualising, querying and analysing data, ultimately allowing the interaction between different operators/experts, such as Epidemiologists, GIS and Data manager, and decision makers.
Materials and methods
A dedicated Data Warehouse (DWH) infrastructure was developed using RDBMS Oracle®11g to harmonise data from different sources (e.g. spatial data, animal registry, production). The available data and epidemiological information were collected and visualised via a set of web applications, developed using HTML5, JavaScript, PHP/Java technologies, on RDBMS PostGreSQL®. All the official documents, on each outbreak and control measures, were stored using a web-based document and activity flows software (ARXivarNEXT).
Results
The developed SDS provides a useful tool to explore data under different perspectives. Information can be visualised both spatially (webGIS) and in non-spatial ways (tables, charts) through dedicated web applications, thanks to the implementation of the DWH, integrating different types of data from multiple databases. In addition,ArxivarNEXT allows to quickly find and retrieve all the formal documents.
Conclusion
The proposed architecture overcomes the weakness of a closely coupled design, enabling dynamic data integration from different providers, allowing interoperability between data and application. The SDS for the management of the HPAI epidemic has proved its helpfulness in decision making,
showing the real time progression of the disease spread.
Talk at the 16th International Symposium of Veterinary Epidemiology and Economics, Halifax, Canada (7-12 August, 2022)
The loop sequence of predicted RNA stem-loops is a key determinant driving insertions at the hemagglutinin cleavage site: a potential explanation for highly pathogenic avian influenza virus emergence

M.I. Spronken, M. Funk, A.P. Gultyaev, A.C.M. de Bruin, R.A.M. Fouchier, M.R. Richard
Abstract
Upon introduction of H5 or H7 low pathogenic avian influenza viruses (LPAIV) into poultry, highly pathogenic viruses (HPAIV) may emerge, resulting in severe disease with mortality rates of up to 100%. The LPAIV to HPAIV transition is the result of the acquisition of a multi-basic cleavage site (MBCS) in the hemagglutinin (HA). HAs with an MBCS are cleaved by ubiquitous furin-like proteases, supporting systemic viral dissemination in poultry. The exact molecular mechanism underlying this genetic change and why MBCS acquisitions have been exclusively observed in H5 and H7 subtypes remain unknown. We developed a novel, high-throughput and sensitive system with which insertions at the HA cleavage site are detected. Single nucleotides are deleted in the cleavage site of HA. Upon use of these templates in reverse genetics, viruses with HAs presenting insertions or deletions (indels) to repair the HA frame are under strong positive selection. Predicted RNA stem-loop structures of H5 and H6 HA cleavage site regions were the starting point in understanding the features of H5 HA (RNA structure/sequence) that promote insertions. H6 HA was selected as no H6 viruses have acquired an MBCS in nature so far. Indels were detected in a representative LPAIV H5 cleavage site at a low frequency. Indel frequency was increased when the number of adenines (A) in the loop was increased, even upon only one single nucleotide substitution. In contrast to H5, multiple substitutions to increase the number of As and resemble the H5 loop sequence were required in H6 for indels to be detected, yet at lower frequency than in H5. These data shows that sequence is a key determinant driving insertions at the HA cleavage site HA, potentially explaining why HPAIVs have emerged from H5 but not from H6 LPAIVs.
Talk at the Negative Strand Virus meeting, Braga, Portugal (17 June, 2022)
STRONG NATURAL SELECTION OF HIGHLY PATHOGENIC AVIAN INFLUENZA VIRUS MINOR VARIANTS IN CHICKENS BUT NOT IN DUCKS

Anja de Bruin, Monique Spronken, Adinda Kok, Miruna Rosu, Dennis de Meulder, Stefan van Nieuwkoop, Pascal Lexmond, Theo Bestebroer, Sander Herfst, Ron Fouchier, and Mathilde Richard
Poster at the Negative Strand Virus meeting, Braga, Portugal (17 June, 2022)
The constant threat of bird flu viruses to humans and farmed birds

Thijs Kuiken
Presentation for EU Parliament Intergroup on the welfare and conservation of animals, Brussel, Belgium (24 March, 2022)
Addressing avian influenza and other health hazards in poultry production according to the One Health approach

Thijs Kuiken
Talk at the EPIC conference, Edinburgh, UK (01 March, 2022)
When Avian influenza takes flight


Mariëlle van Toor & Thijs Kuiken
Talks at the Netherlands Centre for One Health webinar series, online (24 February, 2022)
Virulence determinants of clade 2.3.4.4 H5N8 viruses in Pekin ducks

David Scheibner, Angele Breithaupt, Christine Luttermann, Diana Palme, Maryna Kuryshko, Claudia Blaurock, Thomas C. Mettenleiter and Elsayed M. Abdelwhab
Abstract
Background
Avian influenza viruses (AIV) are a common threat to poultry and human health and cause periodically huge socioeconomic losses worldwide. Depending on the two major surface glycoproteins, 16 hemagglutinin (HA) and 9 neuraminidase (NA) distinct subtypes can be distinguished. All HA/NA subtypes are circulating in wild waterfowl, the natural reservoirs. In domestic birds, AIV are either low pathogenic (LPAIV) causing mild or no clinical signs or highly pathogenic (HP) causing up to 100% mortality in few days. In chickens, LPAIV H5 and H7 viruses can evolve to HPAIV due to acquisition of polybasic amino acids in the HA cleavage site (HACS). Conversely, ducks rarely succumb to morbidity and mortality after HPAIV infections and the HACS alone is not the main virulence determinant. Since 2014, H5N8 clade 2.3.4.4 caused high mortality in domestic and wild birds worldwide in three major waves in 2014, 2016 and 2020/2021. Although all H5N8 viruses possessed polybasic HACS and were highly virulent in chickens, viruses in 2016-2021 only were high pathogenic in Pekin ducks. In this study, the virulence determinants leading to differences in virus phenotypes between German H5N8 clade 2.3.4.4 viruses in 2014 and 2016 were analyzed.
Methods
Sequence analysis of H5N8 viruses from 2014 and 2016 revealed several mutations in all eight gene segments. Using reverse genetics, single, double and multiple H5N8-2016 segments were swapped with those from H5N8-2014 and recombinant viruses were rescued and characterized. The minimal genetic constellation for the exhibition of high virulence in Pekin ducks was determined.
Results
In vivo, the H5N8-2016 HA alone did not increase the virulence of H5N8-2014 in Pekin ducks. We further tested double reassortants of H5N8-2014 carrying HA in addition to single segments from H5N8-2016. Virulence of H5N8-2014 increased dramatically after reassortment of H5N8-2016 HA in combination with NP, NS and/or NA. The high mortality caused by these viruses was associated with the degree of endothelial tropism and systemic spread in different tissues. In vitro, the replication of the H5N8-2014 virus in primary duck cells was increased by swapping the HA and NP from H5N8-2016. The latter exhibited increased HA receptor binding affinity and NA activity compared to the H5N8-2014 virus.
Conclusion
Taken together, in this study we determined the multifactorial genetic constellation and underlying mechanism for the high virulence of H5N8 clade 2.3.4.4 viruses in Pekin ducks, which improves our current understanding for the evolution of HPAIV in different avian species.
Talk at the 8th ESWI Influenza Conference, virtual (4 - 7 December 2021)
Genetic determinants for virulence and adaptation of clade 2.3.4.4 H5N8 viruses in chickens, ducks and mammals

Elsayed M. Abdelwhab, Claudia Blaurock, David Scheibner, Angele Breithaupt, and Thomas C. Mettenleiter
Abstract
Background
Since 1997, avian influenza viruses (AIV) of H5Nx goose/Guangdong lineage continue to cause severe losses in poultry and wild birds and pose a serious pandemic risk. This H5Nx lineage exhibited high flexibility for mutations resulting in tens of clades/subclades and undergone several reassortment events. From 2014 to 2021, the panzootic H5N8 clade 2.3.4.4 AIV spread on an unprecedented scale to wild and domestic birds worldwide. Compared to the parent clade 2.3.4.4A viruses (designated H5N8-A) in 2013/2015, viruses in clade 2.3.4.4B (designated H5N8-B) from 2015 to 2021 caused high mortality in wild birds and Pekin ducks and was isolated recently from humans and dead seals. Therefore, there is an urgent need to keep vigilance to assess periodically the virulence and transmissibility of H5N8-B in different animals and understand the molecular mechanisms underlying potential virus adaptation. In this comprehensive study, we determined the pathogenicity and virulence markers of H5N8-A and H5N8-B in chickens, ducks and mice and determined the genetic markers for the efficient transmission of H5N8 viruses in Pekin ducks and chickens.
Methods
Recombinant H5N8-A, H5N8-B and H5N8-A viruses carrying gene segment(s) from H5N8-B were successfully constructed using reverse genetics. Replication efficiency in avian and mammalian cells, receptor-binding affinity, sialidase and polymerase activities, interferon (IFN)-antagonism and apoptosis induction were assessed in vitro. In vivo, chickens and ducks (n= 10 each) were inoculated oculonasal with recombinant viruses and one-day post-inoculation five birds were added to assess chicken-to-chicken or duck-to-duck transmission, respectively. Morbidity, mortality, virus excretion, tissue distribution and histopathological alterations were described. Furthermore, in mice, we assessed the impact of selected recombinant H5N8-B viruses on bodyweight gain, mortality, virus replication in the lungs and interferon induction.
Results
In vitro, compared to H5N8-A,H5N8-B virus exhibited increased binding affinity to avian-sialic acid receptors and higher NA activity. In duck cells, but not in chicken or human cells, H5N8-B replicated at higher levels than H5N8-A. Both H5N8-A and H5N8-B blocked IFN-β induction efficiently despite of the striking preferential differences in the NS1 C-terminus domain (CTE). In chickens, both viruses were highly virulent, while H5N8-B was more virulent in Pekin ducks and mice than H5N8-A. In ducks, HA, NA and NS played a role in H5N8-B virulence and transmission. Mutations in the NS1 CTE of H5N8-B (i) reduced virus shedding, transmission and/or endothelioptropism in chickens and ducks, (ii) reduced virulence and replication in mice and (iii) contributed to the high efficiency of H5N8-B for blocking IFN and apoptosis inductions in human lung cells indicating a role in virus adaptation in mammals.
Conclusion
The recent H5N8-B virus exhibited high virulence in chickens, Pekin ducks and mice compared to the predecessor H5N8-A virus. NS segment contributes to H5N8-B fitness in birds and mammals.
Talk at the 8th ESWI Influenza Conference, virtual (4 - 7 December 2021)
Adaptive potential of zoonotic avian influenza H7N9 virus to ducks

Susanne Koethe, Lorenz Ulrich, Angele Breithaupt, Christian Grund, Anne Pohlmann, Timm Harder and Martin Beer
Abstract
Migratory waterfowl are potential vectors for the long-range transmission of avian influenza viruses (AIV) globally. In 2013, low pathogenic avian influenza viruses (LPAIV) of subtype H7N9 emerged in China and evolved into highly pathogenic avian influenza virus (HPAIV) variants that were first detected in the beginning of 2017. Extensive co-circulation of both pathotypes was particularly restricted to galliform poultry but caused several thousand severe human infections with a high fatality ratio in China. The systematic surveillance revealed constant evolution of AIV H7N9 and reassortment events with the Eurasian wild bird gene pool. Spillover and adaptation of either LPAIV or HPAIV H7N9 from galliform to anseriform poultry into migratory waterfowl would justify severe concerns about a possible transcontinental spread of these highly zoonotic viruses.
The potential of AI H7N9 viruses to adapt to wild waterfowl was determined using an LPAIV (A/duck/Japan/AQ-HE28-3/2016) and two HPAIV H7N9 isolates, (A/duck/Japan/AQ-HE29-22/2017 (HE29-22), and (A/duck/Japan/AQ-HE29–52/2017) (HE29-52)). Replication competence and transmissibility, respectively, were characterized in embryonated duck eggs, in one-week old ducklings and in four-week old ducks.
The LPAIV strainrevealed no pathogenicity and transmissibility in ducks. In contrast to the LPAIV, the two HP variants induced systemic infection in duck embryos. When applied intra-muscularly to one-week old ducklings, the HPAIV HE29-22 and HE29-52 isolates caused mortality, indicating increased virulence. In four-week old ducks, oronasal inoculation of both respective HPAIV H7N9 isolates lead to virus shedding and efficient transmission to contact ducks without onset of disease.
The data indicate a significant adaptive potential of HPAIV, but not LPAIV, H7N9 isolates to ducks. Asymptomatic but productive infection in four-week old ducks justifies fears of undetected regional and possibly global virus spread with ducks and migratory anseriform birds, respectively.
Talk at the 8th ESWI Influenza Conference, virtual (4 - 7 December 2021)
Species-specific endotheliotropism of Highly Pathogenic Avian Influenza Virus in avian hosts

Anja C.M. de Bruin, Monique I.J. Spronken, Theo M. Bestebroer, Pascal Lexmond, Zhen Wei Marcus Tong, Michelle Baker,Kirsty R. Short Ron A.M. Fouchier and Mathilde J. Richard
Abstract
Introduction
Highly pathogenic avian influenza viruses (HPAIV) of the subtypes H5 and H7 emerge from low pathogenic precursors upon transmission from wild waterfowl, their reservoir, to poultry. In poultry, HPAIV are responsible for systemic disease with marked endotheliotropism resulting in outbreaks with mortality rates as high as 100%. The HPAIV phenotype is the result of the acquisition of multiple basic amino acids at the cleavage site of the hemagglutinin (HA) protein. This allows for HA activation by ubiquitously expressed proteases which leads to systemic virus dissemination. HPAIV infections in wild birds are, in general, of milder pathogenicity and are not associated with endothelial cell tropism. The involvement of endothelium in immunopathogenesis is well established in mammalian influenza infections, but data from avian species is lacking. This study investigates virus replication, in relation to protease expression, and immune responses in chicken and duck endothelial cells which could contribute to observed differences in HPAIV pathogenesis.
Methods
Primary endothelial cell cultures of both duck and chicken origin were established, in which viral growth kinetics and host responses were compared. Expression of HA-activating proteases was analyzed by PCR followed by functional characterization in an overexpression system. A transcriptomics approach was applied to HPAIV-infected cells to study host responses and the production of inflammatory cytokines was studied during infection and after Toll-like receptor stimulation.
Results
Surprisingly, the amount of produced infectious virions upon HPAIV infection was comparable between primary chicken and duck endothelial cells, showing only a trend towards lower initial infection percentages in duck cells than in chicken cells. Messenger RNA of the HPAIV-activating proteases furin and pc5/6 were expressed in duck endothelial cells and duck furin cleaved HPAIV HA when both proteins were overexpressed in a furin-deficient cell line. Transcriptomic analysis of chicken endothelial cells hinted towards a more pro-inflammatory state, which was supported by high IL-6 and IL-8 mRNA induction upon Poly(I:C) treatment. Duck endothelial cells overexpress genes within pathways related to immune regulation and the type I interferon response.
Conclusion
Primary duck endothelial cells sustained multi-cycle growth of HPAIV which was supported by the expression of adequate HPAIV-activating proteases. Despite comparable levels of virus replication, host responses upon infection differed between chicken and duck endothelial cells. Chicken cells mounted a pro-inflammatory response whereas duck cells increased their antiviral defenses. The lack of endotheliotropism of HPAIV in ducks in vivo is not fully recapitulated within our monoculture experiments. Therefore, we are currently developing an epithelial-endothelial coculture setting to study viral dissemination, barrier integrity, and host responses.
Talk at the 8th ESWI Influenza Conference, virtual, (4 - 7 December 2021)
Aviaire influenza vanuit een One Health benadering (Avian influenza from the One Health approach)

Thijs Kuiken
Talk at the Dutch National Zoonoses Symposium (25 November 2021)
Variable susceptibility of poultry species to H7N7 avian influenza viruses is host response dependent
Shannon Leetham, Alexander MP Byrne, Joe James, Saumya S Thomas, Paul Skinner, Felicity Wynne, Benjamin Mollett, Alejandro Núñez, Marek J Slomka, Ashley Banyard, Ian H Brown, Falko Steinbach, and Sharon M Brookes
Abstract
Avian Influenza viruses (AIVs) infect domestic poultry causing a spectrum of disease broadly defined as either low pathogenicity (LPAIV) or high pathogenicity (HPAIV). H5 and H7 AIV subtypes can mutate from LPAIV to HPAIV, via mutation within the viral haemagglutinin protein cleavage site, causing outbreaks which are characterised by high morbidity and mortality. This mutation is a key virulence determinant, but other factors, including host immunological responses, are believed to play a significant role in disease status. A key knowledge gap following AIV infection is the role of the host innate and adaptive immune responses and their contribution to disease outcome. To investigate this, the differences in host immune response genes to two H7N7 LPAIV/HPAIV pairs was assessed in vivo in naïve specified-pathogen free chickens. The LPAIV/HPAIV pairs selected for this work, cause outbreaks in Europe in 2015, in the UK and Germany. For each virus, 18 chickens were infected with 106 EID50 and then culled at 24-, 36- and 48-hours post-infection (hpi), along with uninfected animals collected at 0 hpi. At each timepoint, a selection of immunological cells and tissues were collected for qPCR analysis. The results from these in vivo studies showed changes in host gene responses between LP and HPAIV infections and this demonstrates that even viruses that are geographically- and temporally-related can cause differential immune responses. However, ongoing work will further elucidate the potential mechanisms and factors that influence these differences, and therefore increase our understanding of how AIV pathotype influence clinical disease in poultry.
Talk at the Microbiology Society Conference; Avian Infectious diseases 2021, virtual, (15-19 September 2021)

DNA barcoding of faecal material as a non-invasive approach to active wild-bird surveillance for notifiable avian diseases
Saumya S Thomas, Amanda H Seekings, Paris Patapiou, Craig S Ross, Ashley C Banyard and Marek J Slomka
Abstract
The utility of DNA barcoding to define biological species has been demonstrated across multiple sectors for both species conservation and disease detection. Notifiable avian diseases (NADs) are a key threat to the poultry sector globally and recent incursions of highly pathogenic (HP) avian influenza viruses (AIVs) have had a significant economic impact in the UK. Wild-birds are responsible for the maintenance and dissemination of AIVs as well as other NAD agents such as avian paramyxoviruses (APMVs), the causative agents of Newcastle disease (ND). HPAIVs (of the H5Nx subtype) have repeatedly emerged across Europe, the Middle East and sub-Saharan Africa, causing economically-significant poultry outbreaks as well as significant wild bird die offs. Due to the complexities of catching and sampling wild-birds species, surveillance during both peacetime and outbreaks is passive, whereby swabs are collected from carcasses for testing. However, recent incursions of H5Nx into the UK has driven the need to develop alternative cost effective strategies for sampling bird populations strategically as an early warning system for the emergence of notifiable avian diseases. Collection and testing of faecal material, using molecular tests, can be used to screen material, and where positive samples are detected, the avian species of origin can be defined by DNA barcoding. The ability to sample faecal material from healthy birds, rather than having to catch and swab birds, enables low-cost evaluation of avian populations that can be asymptomatically infected with NADs. As such, screening faecal material will help define high-risk avian populations for incursion of notifiable diseases.
Talk at the Microbiology Society Conference; Avian Infectious diseases 2021, virtual, (15-19 September 2021)

Introduction routes for Newcastle disease virus and highly pathogenic avian influenza virus (H5N8) in a high-biosecurity poultry setting
CJ Warren, MJ Slomka, S Thomas, S Mahmood, J James, AMP Byrne, S Watson, A Nunez, FZX Lean, CS Ross, SM Brookes, AC Banyard, R Hansen, IH Brown
Abstract
Understanding the potential routes for incursion of notifiable avian diseases (NAD) into high-biosecurity poultry settings is critical in enabling mitigation during NAD outbreaks. Recent UK outbreaks demonstrated that bio-secure facilities are readily breached but the route of incursion is often undefined. We established in vivo models to evaluate incursion of NAD in water and feed matrices. To mimic natural incursion, five groups (1-5) of naïve chickens (n=6/group) were housed in separate pens and exposed to virus-contaminated matrices for 24 hours. Matrices included drinking water contaminated with high levels (106 EID50/ml) of NDV-2013 (Genotype VII) (1) and H5N8-2016 (2), with a third group (3) exposed to a lower contamination level (104 EID50/ml). Groups 4 and 5 were exposed to feedcrumb contaminated with H5N8-2016 at either a high- or lower-dose. Two additional groups (6 and 7) were infected via intra-nasal route with high doses of NDV-2013 (6) and H5N8-2016 (7), as positive control birds. Both control chicken groups (6 and 7) succumbed to infection with 100% mortality. High-level water contamination with NDV-2013 (1) resulted in 100% mortality, whilst the corresponding experiment with H5N8-2016 (2) resulted in 67% mortality. All birds exposed to lower dose H5N8-2016 in water (3), or feedcrumb contaminated with high-dose (4) or lower-dose H5N8-2016 (5) remained uninfected and no birds seroconverted. These data suggest differences exist in virus bioavailability from water and feedcrumb matrices and that virus contaminated water represents a highrisk viral incursion route with onward transmission potential.
Talk at the Microbiology Society Conference; Avian Infectious diseases 2021, virtual, (15-19 September 2021)

The interplay between avian influenza viruses and their hosts: early transcriptome response of galliformes infected with H7 strains showing different pathogenic potential

Gianpiero Zamperin, Alice Bianco, Jacqueline Smith, Alessio Bortolami, Lonneke Vervelde, Alessia Schivo, Andrea Fortin, Sabrina Marciano, Valentina Panzarin, Eva Mazzetto, Adelaide Milani, Yohannes Berhane, Paul Digard, Francesco Bonfante, Isabella Monne
Abstract
Influenza A viruses (AIVs) are important veterinary and human health pathogens and avian species represent their natural hosts. Many subtypes of AIVs exist, each characterized by a specific pair of surface glycoproteins, the hemagglutinin (HA) and the neuraminidase (NA). AIVs infecting poultry can be divided into two distinct groups on the basis of the severity of the disease they cause. Once low pathogenic avian influenza (LPAI) viruses of the H5 and H7 subtypes from wild birds enter into poultry species, there is the possibility of them mutating to highly pathogenic avian influenza (HPAI). This mutation results in severe epizootics with up to 100% mortality, with major economic impact on poultry production. It follows that LPAI infections with viruses of the H5 and H7 subtypes are of great concern. It is still unclear why the H5 and H7 subtypes are more prone to evolve into HPAI forms than any other AIV HA subtypes. Although AIVs are inextricably linked to their hosts in their evolutionary history, the contribution of host-related factors in the emergence of HPAI viruses has only been marginally explored so far. In order to close such gap, we studied the early stages of transcriptome response in the tracheal tissue of chickens infected with two pairs of H7 strains which demonstrated a distinct ability to evolve their pathotype in the field. Our results show that H7 precursors of HP viruses trigger the host immune response, while in chickens infected with the H7 strains that do not mutate to the HP phenotype, the majority of differentially expressed genes were involved in metabolic and cellular division processes, with very low involvement of immunity genes. These findings suggest that, in the fight between host and virus, the immune response may represent a selective factor favoring the mutation of influenza viruses themselves.
Poster at the MEEGID XV - 15th International Conference on Molecular Epidemiology and Evolutionary Genetics of Infectious Diseases (2 - 5 November 2021)
Hoog-pathogene vogelgriep in 2021 volgens de One Health benadering (Highly pathogenic bird flu in 2021 according to the One Health approach)


Thijs Kuiken
Presentation in the zoo "Ouwehands", Rhenen (23 October 2021)
Influenza

Mathilde Richard
Virology course (4 - 8 October 2021)
Investigating differential expression in avian innate immune responses to respiratory viruses
Samantha Sives, Dominika Borowska, Karen Bryson, Kate Sutton and Lonneke Vervelde
Abstract
Our understanding of the avian respiratory innate immune response to viruses is limited and often hindered by genetic diversity from dual aspects of both hosts and viruses. Development of sensitive high-throughput transcriptomic assays for analysing these innate responses is of paramount importance to improve understanding of the complex interplay between viruses and the avian respiratory immune system. We have used an in house developed multiplex qRT-PCR Fluidigm Dynamic Array (PMID 31794562) to study transcription and phenotypic analysis of chicken innate immune-related genes. The multiplex qPCR included a wide array of genes induced by activation of the innate immune response, including type 1 IFNs, IFN-signalling pathways, IFN-stimulated genes, cytokines, chemokines and pathogen-recognition receptors. Here, we investigated the differences and commonalities in respiratory immune responses between avian viruses with differing levels of attenuation and pathogenicity. Utilising a novel combination of: (1) in vitro primary lung macrophages infected with Infectious Bronchitis virus (IBV), Avian Influenza virus (AIV) or Newcastle Disease virus (NDV), (2) ex vivo-derived precision cut lung slices infected with AIV and (3) an in vivo tissue panel (trachea, lungs) from chickens infected with respiratory or nephropathogenic strains of IBV. Preliminary analysis revealed both similarities and differences in the respiratory innate response to viruses differing in virulence and pathogenesis. An improved understanding of how viruses infect chickens, the immune genes involved in these interactions and viral immune evasion strategies, is of economic importance for the poultry industry through improved vaccine efficacy and development of novel control methods.
Presentation at the 7th European Veterinary Immunology Workshop (29-31 August 2021)

Sequential Infection of Poultry with Low and then Highly Pathogenic H7N7 Avian Influenza Viruses: Investigating Factors Contributing to Disease Outcome
Alexander Byrne, Shannon Leetham, Joe James, Saumya Thomas, Caroline Warren, Fabian Lean, Alejandro Núñez, Ashley Banyard, Ian Brown, Marek Slomka, Sharon Brookes
Abstract
Factors driving the evolution of high pathogenicity avian influenza viruses (HPAIVs) from low pathogenicity viruses (LPAIVs) remain undefined. In July 2015, a disease investigation prompted by increased mortality on a commercial chicken farm, identified an incursion of H7N7 LPAIV that circulated within birds on the premises shortly before mutating to a HPAIV. HPAIV induced mortality in a subset of chickens, but the majority were protected, potentially due to prior exposure to LPAIV. Whilst genetic evidence for both H7N7 LPAIV and HPAIV were detected during the outbreak, only the HPAIV (A/chicken/England/26352/2015, H7N7-HP) was successfully isolated. To investigate the potential for protective responses being induced by prior LPAIV circulation, eight donor chickens were experimentally infected with A/mallard/Netherlands/19/2015 H7N7 LPAIV (H7N7-LP), a virus closely related to H7N7-HP, before eight contact chickens were introduced at 1 day post-infection (dpi). All eight donor chickens shed virus and seroconverted, whilst 50% of contact chickens shed virus and only 37.5% seroconverted, demonstrating H7N7-LP transmission. At 14 dpi, all 16 donor and contact chickens, alongside eight naïve positive control chickens, were challenged with H7N7-HP. From the control and contact groups, 100% and 87.5% of chickens shed H7N7-HP, with 62.5% and 50% mortality in these groups, respectively. None of the LP-donor chickens shed H7N7-HP or succumbed to infection. This demonstrated that infection of chickens with a LPAIV protects against infection with an antigenically-related HPAIV providing further insights into the clinical outcome during the outbreak. Analysis of these samples may help determine factors involved in LPAIV to HPAIV mutation.
Poster at the Microbiology Society Annual Meeting, virtual, (26-30 April 2021)

Different genetic constellations for high virulence, transmission and tropism of highly pathogenic H7N7 avian influenza virus in turkeys and chickens
David Scheibner, Reiner Ulrich, Olanrewaju I. Fatola, Marcel Gischke, Ahmed Salaheldin, Jutta Veits, Ulrike Blohm, Thomas C. Mettenleiter and Elsayed M. Abdelwhab
Abstract
Highly pathogenic (HP) avian influenza viruses (AIV) cause severe mortality in chickens (Gallus gallus domesticus) and turkeys (Meleagris gallopavo), although turkeys are more sensitive than chickens. Little is known about the virulence determinants in both bird species, beyond the polybasic cleavage site (CS) in the hemagglutinin (HA). In 2015, HPAIV H7N7 and a putative low pathogenic (LP) ancestor were isolated from the same chicken farm. In addition to the polybasic HA CS, mutations in all gene segments were observed. The aim of this study was to investigate the impact of mutations in different segments in addition to the polybasic CS on virulence, transmission, replication and tissue tropism in chickens and turkeys. Using reverse genetics different virus reassortants with an LP backbone carrying the polybasic CS and single or multiple HP segments were generated. Interestingly, viruses carrying the HP NS or M gene segments or NS segment were as virulent and transmissible as the HPAIV in turkeys and chickens, respectively. All viruses were detected in the brain, although the endothelial tropism was exclusively found in chickens where the HACS and to lesser extent NA are major determinants for the endotheliotropism. In vitro characterization revealed comparable replication kinetics of all viruses but lower NA and higher polymerase activity of HP compared to LP. Taken together, this study showed two major differences between chickens and turkeys: more genetic constellations to confer high virulence and transmission in turkeys than in chickens and neurotropism in turkeys vs. endothelial tropism in chickens. M and NS genes were required for widespread distribution and severe inflammation particularly in the brain and lymphatic organs.
Poster at the 30th Annual Meeting of the Society for Virology (24-26 March, 2021)
Keeping pace with the evolution of HPAIV H5 - Characterization of a novel HPAIV H5N8 reassortant 2019/2020
Susanne Koethe, Lorenz Ulrich, Kore Schlottau, Jacqueline King, Angele Breithaupt, Donata Hoffmann, Anne Pohlmann, Christian Grund, Timm Harder, and Martin Beer
Abstract
Novel highly pathogenic avian influenza virus (HPAIV) reassortants (clade 2.3.3.4b) were first detected in a greater white-fronted goose in January 2020. Until the end of March, a total of six cases of infection of domestic poultry and wild birds were confirmed in Germany. Full-genome sequencing revealed that the novel reassortant held six segments from a Eurasian/Asian/African HPAIV H5N8 reassortant and two segments from low pathogenic avian influenza H3N8 subtype viruses recently detected in wild birds in Central Russia (King et al., 2020; doi:10.3390/v12030281).
Migratory wild waterfowl, especially dabbling ducks, may play a pivotal role in the maintenance and dissemination of low pathogenic avian influenza viruses (LPAIV) and HPAIV, typically associated with the absence of clinical signs and low mortalities. The excretion of virus by infected wild waterfowl may lead to habitat contaminations whereby perpetuating infections and enhancing the risk of an introduction into poultry holdings.
To characterize the pathogenicity of the novel reassortant HPAIV H5N8 (A/white-fronted goose/Germany-BB/AI00018/2020) for waterfowl and poultry, five Pekin ducks and five chickens each four-weeks old were inoculated with 1x106 EID50 per bird intranasally. One day post infection (p.i.), two naïve sentinel animals each were co-housed. Oropharygneal and cloacal swabs were taken daily. The animals’ health status was scored daily.
The novel HPAIV H5N8 reassortant revealed the capacity to infect chickens and ducks. In both groups, virus shedding started on day one p.i. and higher virus loads were excreted in oropharyngeal than in cloacal samples. All inoculated chickens had to be euthanized on day 5 p.i. due to a moribund health status. No virus transmission to the two sentinel chickens could be observed. In contrast, virus shedding by inoculated ducks was sufficient to infect sentinels. The ducks had to be euthanized individually from day 4 until day 9 p.i. due to severe neurological disorders. Infectious virus could be re-isolated from brain samples of two selected chickens and ducks, respectively.
The novel HPAIV H5N8 reassortant is avian-adapted with augmented virulence for waterfowl, but also showed a delayed onset of disease with a pronounced neurotropism in chickens and ducks. The tendency for frequent reassortment combined with the enhanced virulence for waterfowl of H5 clade 2.3.4.4 viruses emphasizes the need for continued and close monitoring efforts of wild bird and poultry populations.
Presentation at the 30th Annual Meeting of the Society for Virology (24-26 March, 2021)

One Health evaluation of highly pathogenic avian influenza in the Netherlands, 2020/2021

Thijs Kuiken
Webinar NCOH (28 January 2021)
CAPABILITIES AND CHALLENGES - Key challenges in Animal Health
Sharon Brookes
Presentation at the M3 LEP – Local Enterprise Partnership meeting (22 January 2021)

Species-specific endotheliotropism of Highly Pathogenic Avian Influenza Virus in avian hosts

Anja de Bruin
Talk at the 13th CEIRS meeting, virtual, (11-15 January 2021)
The H5 highly pathogenic avian influenza: understanding the patterns of virus spreading to/from Africa
Alice Fusaro, Bianca Zecchin, Bram Vrancken, Celia Abolnik, Rose Ademun, Yao P. Akpeli, Abdou Alassane, Joseph Adongo Awuni, Emmanuel Couacy-Hymann, M’Bétiégué Coulibaly, Emilie Go-Maro, Tony Joannis, Simon Dikmu JUMBO, Germaine Minoungou, Clement Meseko, SOULEY Maman Moutari , Deo Birungi Ndumu, Augustin Twabela, Abel WADE, Lidewij Wiersma, Gianpiero Zamperin, Adelaide Milani, Philippe Lemey, Isabella Monne
Abstract
During the 2006, 2014 and 2016 intercontinental epidemic waves of the H5 highly pathogenic avian influenza virus (HPAIV) of the Gs/GD lineage, the African continent was reached by three distinct H5 genetic clades, namely 2.2, 2.3.2.1c and 2.3.4.4. Our purpose is to investigate the role of Africa in the global spread of the HPAIV H5 and to shed light on the contributions of different avian host populations to the virus introduction and dissemination.
We generated two datasets of the hemagglutinin gene for each clade, one including representative sequences from affected regions throughout the world and from different host species, and the other comprising all the African sequences generated in this study or retrieved from public databases. Spatiotemporal data analysis of the epidemic spread and of host switching patterns were performed through Bayesian phylogeographic analyses in both discrete and continuous space for each dataset using the BEAST v1.8.4 program.
We recognized multiple introductions as well as varying paths of avian influenza incursions into the African continent during the three epidemic waves and were able to demonstrate that Africa had mainly served as an ecological sink of the H5 Gs/GD HPAIV. Within Africa, West Africa acted as a crucial hotspot for virus introduction and dissemination into the continent. A joint analysis of host dynamics and continuous spatial diffusion indicated that both migratory birds and live poultry trade may have played an important role in the spread of the virus into the continent.
Overall, this study suggests that viral sources are not stable over time in the African continent, but can change at each epidemic wave. Enforcing target surveillance in the regions identified as at high risk of virus incursion, such as West Africa, is recommended.
Poster at the Epidemics7 - International Conference on Infectious Disease Dynamics in Charleston, SC, USA (3-6 December 2019)

Using Sediment, Environmental and Wild Bird Samples as Tools for Avian Influenza Virus Surveillance
Alexander MP Byrne, Chelsea G Himsworth, Caroline J Warren, Saumya S Thomas, Natalie Prystajecky, Adam Brouwer, Jun Duan, Marek J Slomka, Rowena Hansen, Michelle Coombe, William Hsiao, Ian H Brown, and Sharon M Brookes
Abstract
Multiple global clade 2.3.4.4 H5Nx highly pathogenic avian influenza viruses (HPAIVs) have occurred since 2014, typified by circulation in wild birds and poultry outbreaks. Surveillance in the UK for such threatening avian influenza viruses (AIVs) that have the potential to cause significant, has included a risk-based strategy for wild birds, whereby clinical samples from carcasses are laboratory screened for H5Nx AIVs. Assessment involves initial testing by generic real-time reverse transcriptase PCRs (RRT-PCR), followed by a H5-subtype-specific RRT-PCR, viral RNA sequencing and virus isolation for non-negative samples. However, AIVs are mainly excreted from wild birds via faeces, therefore analysis of environmental samples containing faecal matter may complement testing of wild bird carcasses. The results generated from testing environmental samples collected from studies of experimentally-infected poultry, as well as AIV outbreaks will be described. A pilot study to collect environmental samples from wetland sites where AIV-positive wild birds have been detected previously, has also begun via the Horizon 2020 Delta-Flu project. We have also investigated using a genomics-based approach (targeted resequencing) to detect and characterise AIVs from sediment samples collected during the 2014/2015 H5N2 clade 2.3.4.4 HPAIV outbreak in the Fraser Valley, Canada. Of the 300 sediment samples collected, 20.6% were positive for AIV, which included 13 haemagglutinin and 9 neuraminidase subtypes. It is anticipated that data generated from testing environmental samples may supplement the information that is obtained from wild bird samples, regarding the AIVs that are present in the wild bird environment.
Talk at the EPIZONE Annual Meeting 2019 in Berlin, Germany (26-28 August 2019)

Modulation of highly pathogenic avian influenza virus H5N8 infection in naturally low pathogenic avian influenza virus - exposed mallard ducks
S Koethe, L Ulrich, A. Globig, T Harder, FJ Conraths, M Beer
Abstract
The 2016/2017 highly pathogenic avian influenza virus (HPAIV) H5Nx clade 2.3.4.4b outbreak was the most severe ever reported in Germany with high mortality rates not only among domestic birds, but also in wild birds.
Mallard ducks have been kept as sentinels for circulating field AIV in an aviary located at the shallows of the Baltic coast in close contact to wild waterfowl and migratory birds. The mallards were regularly tested for AIV infection. Between August 2017 and October 2018, four low pathogenic (LP) AIV infections of individual mallards were confirmed - H1N3, H3N8, H4N6 and HxN9. One month prior to the HPAIV study, all mallard ducks were tested seropositive in an IAV NP-ELISA, and two out of seven had H5-specific antibodies.
To study dynamics of HPAIV H5N8 clade 2.3.4.4b infection after a prior natural field exposure to LPAIV, seven LPAIV exposed mallards were challenged with HPAIV H5N8. The surviving ducks were re-challenged with the homologous HPAIV H5N8. For both challenge infections, virus transmission to seronegative juvenile Pekin contact ducks was investigated. Seronegative adult Pekin ducks served as a control group.
Summarizing the results, during the first challenge infection all control group ducks died peracutely or had to be euthanized due to severe neurological symptoms. However, although one mallard duck died, the mallards showed only very few clinical symptoms, but three out of four contact juvenile Pekin ducks died. H5-specific antibodies were detectable in all surviving birds. Infectious virus could be isolated from several pond and drinking water samples. In contrast, after the homologous H5N8 re-challenge, neither inoculated nor contact ducks showed any clinical symptoms, nor was an AI-specific antibody titer increase or seroconversion of contact animals determined.
In conclusion, the study simulated an HPAIV H5N8 outbreak under semi-natural conditions with LPAIV-exposed waterfowl. Mallards with preceding LPAIV immunity, did not show any severe clinical or fatal disease, but sufficient virus shedding caused fatal illness in naïve contact ducks. In contrast, H5N8-convalescent animals developed a very robust immunity when re-challenged with the homologous virus shown by the absence of virus shedding and seroconversion of the contact animals.
Talk at the European Congress of Virology in Rotterdam, The Netherlands (28 April - 1 May 2019)

Impact of the polybasic cleavage site within the HA of a recent German H7N7 virus on its pathogenicity in different poultry species
Scheibner D., Veits J., Mettenleiter T.C. and Abdelwhab E.M.
Abstract
Virulence of avian influenza viruses (AIVs) is largely dependent on the amino acid sequence of the hemagglutinin cleavage site (CS). Low pathogenic AIV (LPAIV) carrying an HA with a monobasic CS which is activated by trypsin-like proteases in the respiratory and intestinal tracts cause only local infections with mild clinical signs, if any. Some H5 and H7 subtypes exhibit high pathogenicity (HP) by acquisition of a polybasic CS after circulation of LP precursors in terrestrial poultry. The polybasic CS of HPAIV is cleaved by ubiquitous furin-like proteases causing systemic infections and high mortality. In 2015, in an outbreak in poultry in Germany LP and HP H7N7 viruses could be isolated on the same farm indicating precursor-progeny relationship. Here, we investigated the pathogenicity of these LP and HP viruses in chickens, Muscovy ducks and turkeys via oculonasal and intravenous inoculation and analyzed virulence determinants by reverse genetics. Muscovy ducks showed no or mild clinical signs, while turkeys and chickens died after infection with HPAIV. Insertion of a polybasic CS into the HA of the LP virus (LP-poly) increased pathogenicity for chickens and turkey drastically, but was not sufficient for 100% mortality. LP-poly was less virulent in turkeys than in chickens exhibiting IVPI values of 2.8 and 1.9, re-spectively. Thus, virulence determinants differ between these two species. Our results contribute to understand the pathobiology and evolution of recent LP and HP H7N7 viruses in different poultry species.
Poster at the 29th Annual Meeting of the German Society for Virology in Düsseldorf, Germany (20-23 March, 2019)

Selective pressures driving the emergence of highly pathogenic influenza virus in avian hosts

Anja de Bruin
Poster at the 23rd Moledular Medicine Day, Rotterdam, (14 March 2019)
Transmission dynamics of highly pathogenic avian influenza virus A (H5N8) in Italy, 2016-2017
Alice Fusaro, Bianca Zecchin, Paolo Mulatti, Gianpiero Zamperin, Alessia Schivo, Silvia Ormelli, Sabrina Marciano, Lebana Bonfanti, Alessandra Azzolini, Giovanni Cunial, Paola Massi, Anna Moreno, Maria Lucia Mandola, Stefano Marangon, Calogero Terregino, Isabella Monne
Abstract
Between December 2016 and December 2017, Italy was affected by Highly Pathogenic Avian Influenza virus (HPAIV) H5N8 outbreaks both in domestic and in wild birds. The first epidemic wave lasted for six months, from December 2016 to May 2017, with 16 outbreaks in poultry farms and 7 in migratory birds. The second epidemic wave began on the third week of July 2017 and continued until mid-December of the same year. Sixty-seven outbreaks were observed in poultry farms, while only seven cases were reported in wild birds.
To shed light on the source of the Italian H5N8 outbreaks and to investigate the inter- and intra-farm genetic diversity of the circulating viruses, we used a next generation sequencing approach to characterize the complete genome of 115 samples collected from multiple hosts both from each infected farm and from viruses identified in wild species.
Maximum-likelihood phylogenetic trees of the eight gene segments indicated four distinct introductions of AIV genotypes at the beginning of the epidemic (December 2016-February 2017). Since March 2017 one single genotype (H5N8-A/wild duck/Poland/82A/2016-like) had been identified, with the exception of a single virus detected in a turkey farm in October, which turned out to be a reassortant virus for the NP and PA genes, likely acquired from low pathogenic H9 viruses circulating in resident wild birds. Evolutionary and phylogeographic analyses performed using the BEAST v1.8.4 package suggested that genotype H5N8-A/wild duck/Poland/82A/2016-like had further evolved into two distinct clusters, namely Italy-A and Italy-B. Italy-A had probably emerged between February-April 2017 and circulated in the north-east of Italy; differently, Italy-B seems to have appeared between March-July 2017 and mainly spread in the north-western regions.
During the second epidemic wave, epidemiological investigations identified four clusters of secondary cases, involving 36 infected farms. Viruses isolated from each of these clusters grouped together in the phylogenetic trees of all the eight AIV genes and in our median-joining (MJ) network of the eight concatenated gene segments, thus supporting the findings of the epidemiological analysis.
In addition, we demonstrated a high intra-farm genetic diversity (0-9 nucleotide substitutions/genome). This highlighted the importance of genetically characterizing viruses from multiple hosts within a single farm, so as to correctly reconstruct the evolution and the transmission dynamics of an AI epidemic.
Overall these results proved to be instrumental to help epidemiological investigations to discriminate between new introductions and lateral spreads. This type of analyses could be performed in almost a real-time fashion, providing quickly accessible information to generate and/or corroborate hypotheses on the likely epidemiologic pattern of contacts between cases, in order to adjust AI control and prevention policies.
Talk at the 2nd National Congress of the Italian Society for Virology in Rome, Italy (28-30 November 2018)

Investigating Africa’s contribution to the global spread of H5Nx highly pathogenic avian influenza viruses
Alice Fusaro, Bianca Zecchin, Bram Vrancken, Celia Abolnik, Rose Ademun, Yao P. Akpeli, Abdou Alassane, Joseph Adongo Awuni, Emmanuel Couacy-Hymann, M’Bétiégué Coulibaly, Emilie Go-Maro, Tony Joannis, Simon Dikmu JUMBO, Germaine Minoungou, Clement Meseko, SOULEY Maman Moutari , Deo Birungi Ndumu, Augustin Twabela, Abel WADE, Lidewij Wiersma, Gianpiero Zamperin, Adelaide Milani, Philippe Lemey, Isabella Monne
Abstract
In the last two decades, four intercontinental epidemic waves of the highly pathogenic avian influenza virus (HPAIV) of the A/goose/Guangdong/1/1996 lineage have been reported. The worldwide spread of this strain has caused important damages to the poultry industry, but it also represents a serious concern for public health since the virus can be occasionally transmitted to humans. The African continent has been reached by three of these four waves, which resulted in the introduction of three distinct genetic clades, namely clade 2.2 in 2006, clade 2.3.2.1c in 2014 and clade 2.3.4.4 in 2016. The virus has become endemic in poultry in some African regions, such as Egypt and West Africa, where multiple clades are currently co-circulating, creating the opportunity of genetic reassortment and of emergence of viruses with unknown biological properties. To shed light on the potential risk of virus spread from Africa to the other continents, we compared the global HPAIV H5 transmission patterns of these clades and explored the contributions of different avian host populations to virus dissemination.
For each of the three clades, we generated a dataset which included representative sequences from several affected regions throughout the world and from different host species. Based on the spatial distribution and host origin of the available sequences, we identified 9 discrete regions - West Europe, East Europe, Middle East, East Asia, North-Central Asia, South Asia, West Africa, Central-east Africa and South Africa - and 4 host traits - domestic Galliformes, domestic Anseriformes, wild Anseriformes and other wild bird species. We reconstructed the history of epidemic spreads in space and time simultaneously with the host switching patterns through Bayesian phylogeographic analyses in both discrete and continuous space for each dataset using the BEAST v1.8.4 program.
We identified multiple introductions of clade 2.2 from Europe and of clade 2.3.4.4-B from South and North-Central Asia into the African continent, while a single virus spread from South Asia to Africa seems to have been responsible of the 2.3.2.1c incursion. A joint analysis of host dynamics and continuous spatial diffusion indicates that the incursion of the H5 clades into Africa has been driven by wild Anseriformes and domestic Galliformes hosts, thus suggesting that both migratory birds and live poultry trade may have played an important role in the spread of the virus into Africa. Our results identify only few virus migrations from Africa to the Middle East, likely through poultry trade, while no virus movement from Africa to other continents has been observed.
Overall, this study suggests that Africa mainly serves as a sink of the virus. However, the routes of virus introduction into Africa by means of both wild and domestic birds may change at each epidemic wave, making it difficult to predict the source for the next incursion.
Poster at the 2nd National Congress of the Italian Society for Virology in Rome, Italy (28-30 November 2018)

Global Origins Of African Highly Pathogenic Avian Influenza H5Nx Viruses And Intracontinental Spread
Alice Fusaro, Bianca Zecchin, Bram Vrancken, Celia Abolnik, Rose Ademun, Yao P. Akpeli, Abdou Alassane, Joseph Adongo Awuni, Emmanuel Couacy-Hymann, M’Bétiégué Coulibaly, Emilie Go-Maro, Tony Joannis, Simon Dikmu JUMBO, Germaine Minoungou, Clement Meseko, SOULEY Maman Moutari , Deo Birungi Ndumu, Augustin Twabela, Abel WADE, Lidewij Wiersma, Gianpiero Zamperin, Adelaide Milani, Philippe Lemey, Isabella Monne
Abstract
In 2006, 2014 and 2016 Africa was hit by three intercontinental epidemic waves of the highly pathogenic avian influenza virus (HPAIV) belonging to three distinct genetic clades of the A/goose/Guangdong/1/1996 lineage, namely 2.2, 2.3.2.1c and 2.3.4.4, which substantially affected the local poultry industry. In this study we compared the global and intra-African HPAIV H5 transmission patterns of these clades to shed light on the spread of the virus within Africa and to explore the contributions of different avian host populations to virus introduction and dissemination.
For each of the three clades, two datasets of the hemagglutinin gene were generated: one including representative sequences from affected regions throughout the world and from different host species, and one comprising all the African sequences generated in this study or retrieved from public databases. We reconstructed the history of epidemic spread in space and time and the host switching patterns through Bayesian phylogeographic analyses in both discrete and continuous space for each dataset using the BEAST v1.8.4 program.
We identified multiple introductions of clade 2.2 from Europe and of clade 2.3.4.4-B from South and North-Central Asia into the African continent, while a single virus spread from South Asia to Africa seems to have been responsible of the 2.3.2.1c incursion. Our results identify West Africa as the most important area of virus introduction into the continent. A joint analysis of host dynamics and continuous spatial diffusion indicates that the incursion of the H5 clades into Africa is driven by wild Anseriformes and domestic Galliformes hosts, suggesting that both migratory birds and live poultry trade may have played an important role in the spread of the virus into Africa.
This study shows that viral sources are not stable over time in the African continent, but can change at each epidemic wave, making it difficult to predict the source for the next incursion. In addition, our results indicate a strategic role of West Africa in the virus spread within the continent, which may be considered as a hotspot for H5 HPAIV surveillance.
Talk at the International Meeting on Emerging Diseases and Surveillance in Vienna, Austria (9-12 November 2018)

The African continent: an endpoint in the global spread of the highly pathogenic avian influenza H5Nx virus
Alice Fusaro, Bianca Zecchin, Bram Vrancken, Celia Abolnik, Rose Ademun, Yao P. Akpeli, Abdou Alassane, Joseph Adongo Awuni, Emmanuel Couacy-Hymann, M’Bétiégué Coulibaly, Emilie Go-Maro, Tony Joannis, Simon Dikmu JUMBO, Germaine Minoungou, Clement Meseko, SOULEY Maman Moutari , Deo Birungi Ndumu, Augustin Twabela, Abel WADE, Lidewij Wiersma, Gianpiero Zamperin, Adelaide Milani, Philippe Lemey, Isabella Monne
Abstract
In the last two decades, three of the four intercontinental epidemic waves of the highly pathogenic avian influenza virus (HPAIV) of the A/goose/Guangdong/1/1996 lineage reached the African continent, resulting in the introduction of three distinct genetic clades in African poultry, namely clade 2.2 in 2006, clade 2.3.2.1c in 2014 and clade 2.3.4.4 in 2016. To compare the global and within-Africa HPAIV H5 transmission patterns of these clades and to shed light on the contributions of different avian host populations to the virus spread, we performed Bayesian phylogeographic analyses of the hemagglutinin gene of each of the three clades both on a global and local (Africa) scale.
We identified multiple introductions of each clade into the African continent, either with the same (clade 2.2) or a mixed geographic origin (clades 2.3.2.1.c and 2.3.4.4). We find that HPAIV always first arrives in West Africa, followed by spread to Central-East (2.2 and 2.3.4.4) and/or South (2.3.4.4) Africa. A joint analysis of host dynamics and continuous spatial diffusion indicates that the incursion of the H5 clades into Africa is mainly driven by wild Anseriformes hosts, suggesting that migratory birds play a major role in the spread of the virus into Africa. With one exception, we observed no backward spread from Africa to Eurasia.
These findings highlight the strategic role of West Africa, which is located at the intersection of three flyways, as a hotspot for H5 HPAIV surveillance, particularly in wild birds, for which no or very few data are currently available.
Talk at the Virus Genomics and Evolution 2018 in Cambridge, UK (18-20 June 2018)

Genesis of a highly pathogenic virus: what can we learn from deep sequencing data?
Alice Fusaro, Gianpiero Zamperin, Adelaide Milani, Alessia Schivo, Annalisa Salviato, Isabella Monne
Abstract
The mechanism driving the evolution of a low pathogenic (LP) avian influenza (AI) virus into a highly pathogenic (HP) form is currently scarcely understood. Between April and May 2016 Italy reported two H7N7 outbreaks, one in a laying farm (farm-1) and the other in a turkey farm (farm-2). Epidemiological investigations suggested that the LP form of this strain had been introduced into farm-1 through direct contact between wild birds and free-range hens and then rapidly mutated into a HP form within the premise.
To explore the intra-farm evolution of the virus pathogenicity we genetically characterized the H7N7 positive samples collected from symptomatic and asymptomatic animals at farm-1, revealing the co-circulation of both the HP and the LP progenitor virus. A deep sequencing analysis was performed on three LPAI and five HPAI (four from farm-1 and one from farm-2) viruses.
Phylogenetic analyses showed that their genomes clustered together and were separated by long branches from viruses circulating in Eurasia. The LPAI viruses fell at the base of this cluster and possessed a total of 8 nucleotide and 6 amino acid differences compared to the HPAI viruses. Deep sequencing analysis of the eight viruses indicated that the number of genetic differences as well as the complexity of the viral population of the HP viruses from farm-1 was lower compared to the LP viruses (p<0.05), suggesting that the HP viruses had recently experienced a narrow bottleneck.
These results highlight the importance of genetic surveillance within a single farm to monitor the virus evolution.
Poster at the 10th International Symposium on Avian Influenza, Brighton, UK (15 - 18 April 2018)

Strengthening outbreak investigations using a next generation approach
Alice Fusaro, Bianca Zecchin, Gianpiero Zamperin, Alessia Schivo, Angela Salomoni, Annalisa Salviato, Silvia Ormelli, Sabrina Marciano, Lebana Bonfanti, Alessandra Azzolini, Giovanni Cunial, Paolo Mulatti, Calogero Terregino, Isabella Monne
Abstract
A big data approach to assisting the H5N8 highly pathogenic avian influenza (HPAI) outbreak investigations in Italy was used.
To shed light on the source of the Italian H5N8 outbreaks reported since December 2016 and to investigate the inter- and intra-farm genetic diversity of the circulating viruses, we used an ultradeep sequencing approach to characterize the complete genome from multiple hosts collected at each infected farm as well as from viruses identified in wild species, for a total of 115 samples and about 50 gigabases of data.
Maximum-likelihood phylogenetic trees showed that, following the multiple viral introductions that had occurred at the beginning of the epidemic (December 2016-February 2017), a single genetic group seems to have been circulating in Italy. Evolutionary and phylogeographic analyses performed using the BEAST v1.8.4 package indicate that this group has further evolved into two distinct clusters, namely Italy-A and Italy-B. Italy-A probably emerged between February-April 2017 and has been circulating ever since in the north-east of Italy; differently, Italy-B arose between March-July 2017 and has mainly spread in the north-west of Italy. Median-joining network analysis of the inter- and intra-farm genetic diversity highlights the importance to genetically characterize viruses from multiple hosts sampled at each infected farm, so as to correctly reconstruct the evolution and the transmission dynamics of an AI epidemic.
These results were instrumental to discriminate between new introductions and lateral spreads and to demonstrate the potential and challenges of using high-throughput sequencing data for surveillance and control.
Poster at the 10th International Symposium on Avian Influenza, Brighton, UK (15 - 18 April 2018)

Pathogenicity and virulence determinants of recent German H7N7 viruses in different poultry species
Scheibner, D., Salaheldin, A.H., Winter, F., Gischke, M., Veits, J., Mettenleiter, T.C., and Abdelwhab, E.M.
Abstract
Virulence of avian influenza viruses (AIVs) is largely dependent on the amino acid sequence of the hemagglutinin cleavage site (CS). Low pathogenic AIV (LPAIV) carrying an HA with a monobasic CS which is activated by trypsin-like proteases in the respiratory and intestinal tracts cause only local infections with mild clinical signs, if any. Some H5 and H7 subtypes exhibit high pathogenicity (HP) phenotype after acquisition of a polybasic CS after circulation of LP precursors in terrestrial poultry. The polybasic CS of HPAIV is cleaved by ubiquitous furin-like proteases causing systemic infections and high mortality. In 2015, in an outbreak in poultry in Germany LP and HP H7N7 viruses could be isolated on the same farm indicating precursor-progeny relationship. In this study, we investigated the pathogenicity of these LP and HP viruses in chickens, Muscovy ducks and turkeys via oculonasal and intravenous inoculation. Furthermore, virulence determinants of HPAIV were identified using reverse genetics. Muscovy ducks showed no or mild clinical signs, while turkeys and chickens died after the challenge with HP AIV. Insertion of a polybasic CS in the HA of the LP virus (LP_poly) increased pathogenicity for chicken and turkey drastically, but was not sufficient to kill all animals. LP_Poly was less virulent in turkeys than chickens exhibiting IVPI values of 2.8 and 1.9, respectively. Interestingly, LP_poly containing the NS gene from the isogenic HPAIV was as virulent and transmissible as the original HPAIV in chickens. Our results are important to better understand the pathobiology and evolution of recent LP and HP H7N7 viruses in different poultry species.
Talk at the 10th International Symposium on Avian Influenza, Brighton, UK (15 - 18 April 2018)

Survival kinetics of H5N8 HPAI viruses in the environment directly contribute to epidemiological factors in the 2016/17 epizootic
Caroline J Warren, Amanda Seekings, Anita Puranik, Marek J Slomka, Helen Everett, Ian H Brown, Sharon M Brookes
Abstract
Incursion of H5N8 HPAIV in 2014 and re-emergence in 2016 affected the UK poultry sector having severe economic and trade implications, disease free status restored after nine months.
We performed cross-sectional investigations of virus survival of A/wigeon/Wales/052833/2016 (H5N8-2016) and A/duck/England/1279/2014 (H5N8-2014) at 4˚C and 21˚C in neutral media; and modelled survival at 21˚C on plastic and metal surfaces, spray-atomised with virus (10^6 TCID50 /ml). Viral infectivity was quantified using tissue culture (TCID50) as DT, time taken for virus titre to reduce by 1 log10 at temperature (T).
For H5N8-2016 survival investigations, we observed a D4 of 14 and D21 of 6 days respectively; with times to decay to MID50 (ducks) and extinction at 4˚C were 56 and 70 days respectively; at 21˚C were 24 and 28 days respectively. H5N8-2016 showed only short survival on the surfaces tested (D21 <2 hours) and inactivation when 1% Virkon is applied (<10 min). Ongoing work to test H5N8-2014 at two temperatures and in environmental samples will be presented, to compare survival kinetics for the two H5N8 epizootic viruses from 2014/2016.
The data informs risk management, as the susceptibility of emerging avian pathogens to environmental conditions constantly evolves. European winter-spring (4-21˚C) virus survival data and potential maintenance-transmission occurring over 3.5-8 weeks, supports the observed multiple primary introductions from wild birds and secondary spread during the 2016/17 epizootic across Europe.
10th International Symposium on Avian Influenza, Brighton, UK (15 - 18 April 2018)
In vivo modelling of the wild bird / poultry interface: Introduction of H5N6 (2017) HPAIV from infected waterfowl to terrestrial poultry
SS Thomas, AH Seekings, S Mahmood, CJ Warren, A Nuñéz, BC Mollett, J James, C Bianco, MJ Slomka, IH Brown and SM Brookes
Abstract
Clade 2.3.4.4 H5Nx highly pathogenic avian influenza viruses (HPAIVs) of the “goose/Guangdong” lineage have caused epizootics in Eurasia in recent years. Extensive H5N8 wild bird incidents and poultry outbreaks occurred across Europe and the UK during 2016-2017 as a result of wild bird migration. A neuraminidase reassortant event yielded H5N6 in Europe in 2017, with 20 incidents in more than 100 wild birds involving 18 species in the UK between December 2017 and April 2018. Four H5N6 HPAIV poultry outbreaks to date have occurred in continental Europe with no poultry detections in the UK. It is currently unknown whether the current European H5N6 HPAIVs are adapted to spread from wild birds to domestic poultry. We therefore infected three groups of ducks (4-weeks age, n=9 per group) with three different doses of H5N6-2017 (6, 4 and 2 log10 EID50 per duck via the ocular/nasal route) which served as surrogates for infected waterfowl. Each dose group was further divided into three separately-housed subgroups of three ducks each, whereby at 1-day post-infection five ducks, chickens or turkeys were introduced for cohousing with each subgroup. Ducks directly-infected with H5N6-2017 shed virus and transmitted infection successfully to contact ducks, but less efficiently to contact chickens and turkeys. Infrequent transmission to contact chickens and turkeys resulted in typical HPAIV mortality, but also required a longer exposure compared to similar H5N8-2016 trans-species investigations where transmission from directly-infected ducks to terrestrial poultry was much more efficient. Therefore, H5N6-17 is less adapted to readily cross the species barrier from waterfowl to terrestrial poultry than H5N8-16. These in vivo findings have provided evidence which may explain the absence of H5N6 poultry outbreaks in the UK and very limited terrestrial poultry detections in Europe, contrasting with the numerous poultry outbreaks caused by H5N8 HPAIV during 2016-2017.
Oxford, UK (2018)